






About Flexineb
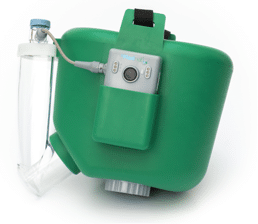
The Flexineb Equine Nebulisation device represents a huge innovative step in administering treatment to the upper and lower respiratory tract. The unique vibrating mesh technology generates a fine mist that has the ability to reach the lowest part of the respiratory treatment, whilst delivering a wide range of solutions such as bronchodilators, corticosteroids, antibiotics, mycolytics and natural therapy solutions.




About Foran
Foran Equine supplements are scientifically formulated by a team of chemists, nutritionists and vets to support your horse's nutritional and performance needs.
About Trindall Equestrian Services Arena Rakes
Arena rakes can be customised to your needs and used on various surfaces. Get in touch with us to discuss your requirements or to learn more about our rakes.
Get in touch
For more information about Flexineb, Foran, Arena Rakes or to ask a question, please get in touch.
